
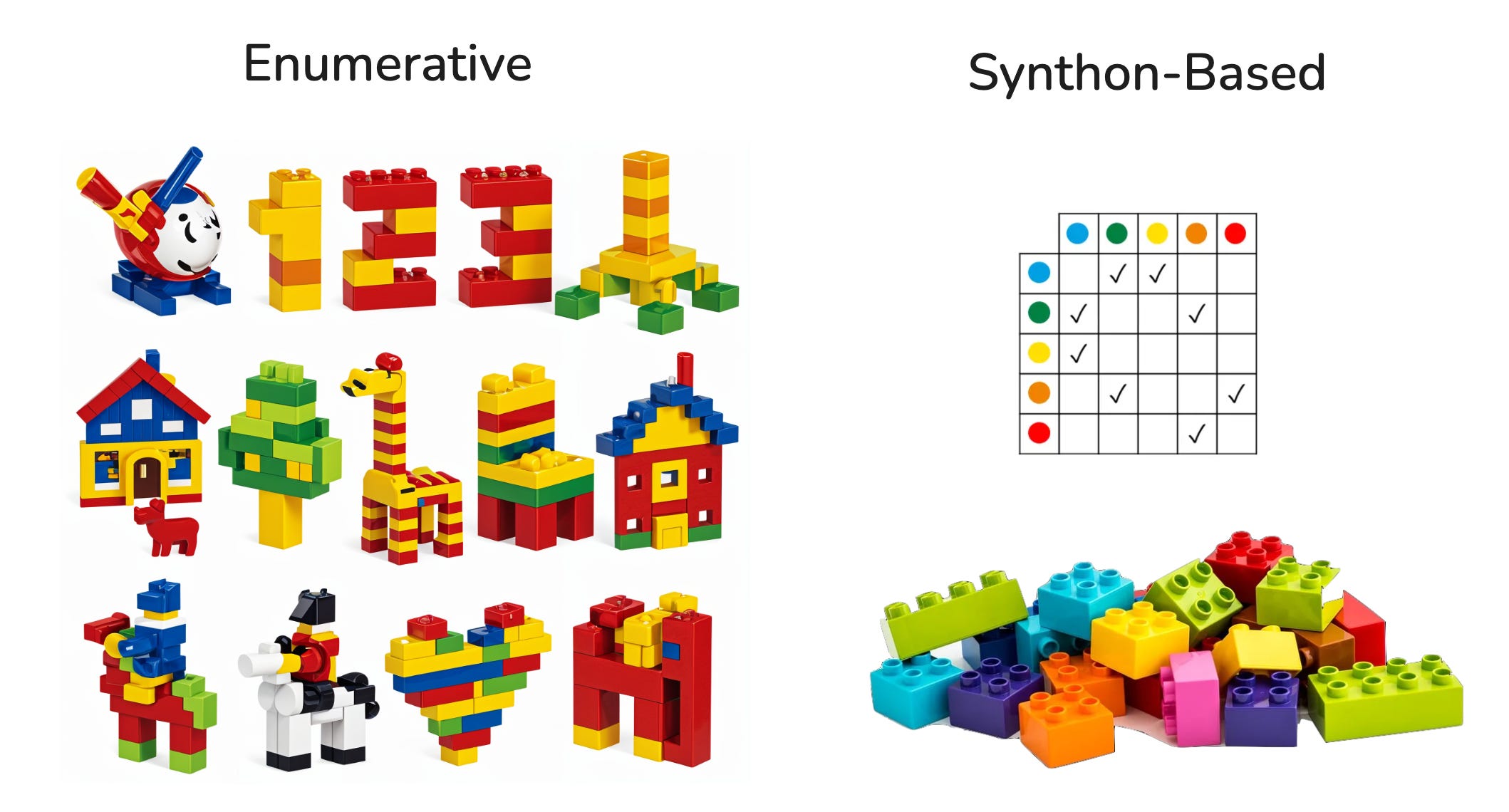
Enumerative vs. Synthon-Based Molecule Search
There are two main approaches to build libraries and navigate in databases of molecules. This post explains enumerative and synthon-based molecular search with intuitive LEGO illustrations.

Modern discovery teams can “search the universe” of purchasable and make-on-demand molecules. Two families of methods dominate:
Enumerative: search over a catalogue where every molecule is known up front.
Synthon-based: search over a virtual space created by combining fragments with reaction rules, instantiating full molecules only for the best candidates.
Both are powerful; both have trade-offs. This post explains what each is, how it works, and when to prefer one over the other—plus a realistic middle ground that often wins in practice.
What is enumerative search?
Plain-English idea: You keep a giant list of molecules and score them directly against your query or target.
Typical inputs
An enumerated library (vendor catalogues, corporate collections, public sets).
A scoring method:
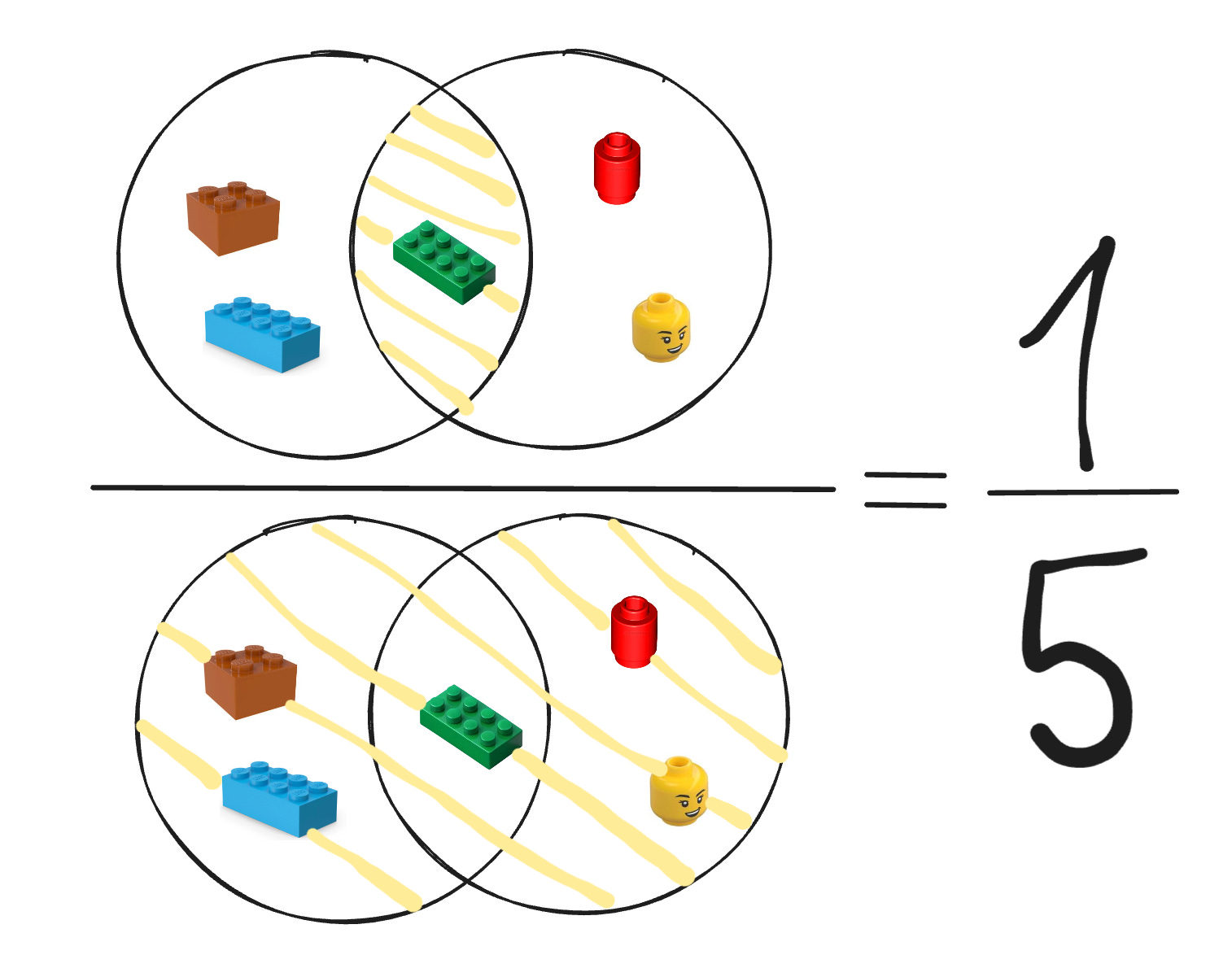
2D: chemical fingerprints (ECFP, path-based, etc.)
3D: shape and electrostatics, pharmacophore/feature overlaps
Embeddings: AI-learned vector representations (CHEESE)
Structure-based: docking or ML surrogates.
Property filters: MW, cLogP, alerts, etc.
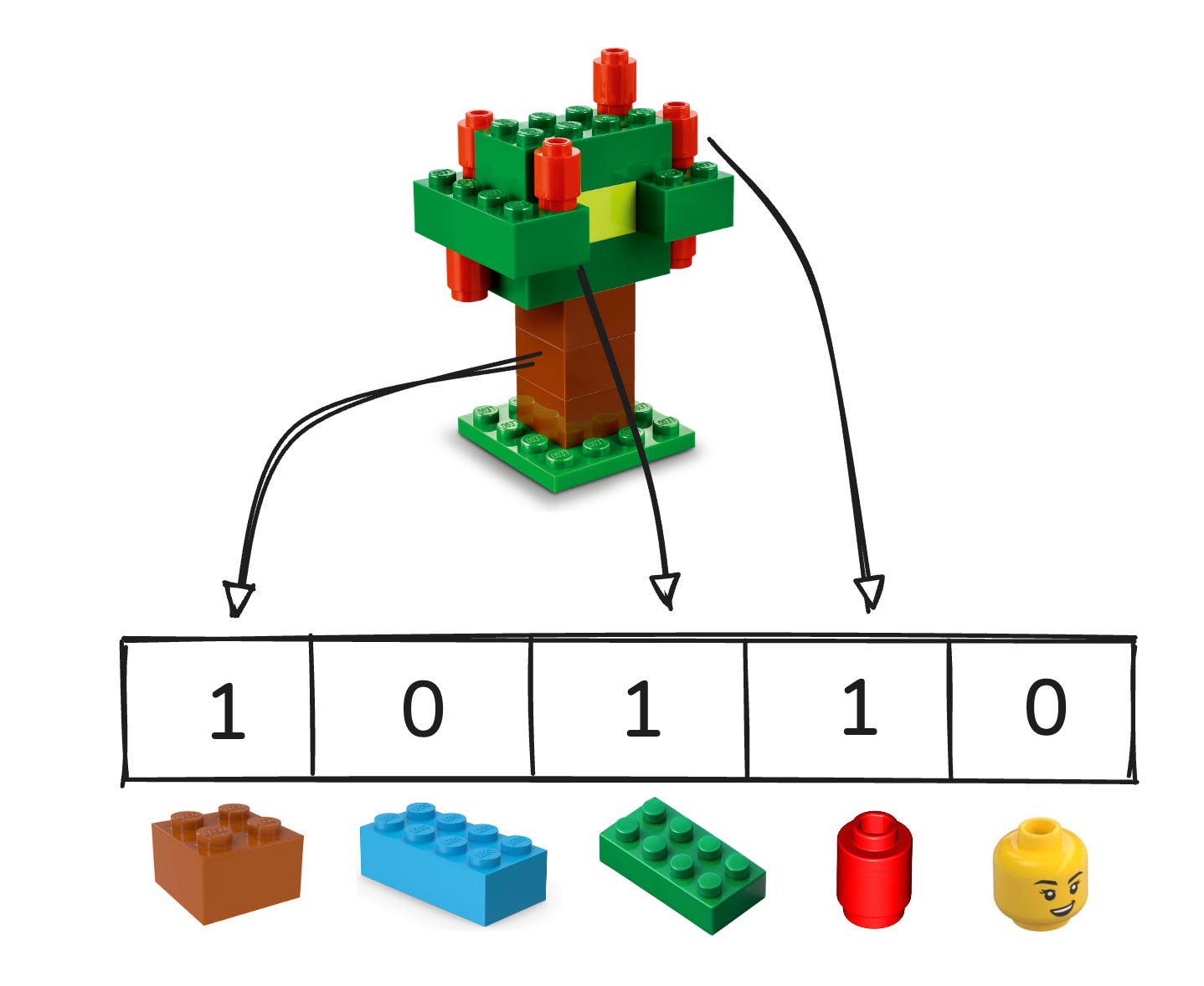
How it works (under the hood)
Each molecule is precomputed to a compact representation
Fast indexes retrieve top candidates without checking every item.
Scores apply to the whole molecule, not fragments.
Strengths
Whole-molecule fidelity. Captures emergent features (shape, electrostatics, stereochemistry, topology) that influence activity.
Actionable hits. Every result is a concrete structure; many are directly purchasable or well-documented synthetically.
Transparent curation. You know exactly what you searched (and what you didn’t).
Limitations
Coverage ceiling. You only find what’s in the list; true novelty is bounded by what’s enumerated.
Cost at scale. Ultra-large sets require serious storage and indexing; rich scores (3D, docking) can be slow or plain impossible on billion scale or larger.
Fingerprint Bias. Standard binary fingerprints are able to capture local patterns and motifs, but not the full shape or complex electrostatics.
What is synthon-based search?
Plain-English idea: Instead of exhaustively listing every possible product, define a chemical space using fragments with attachment points (synthons) and reaction rules, or using feature-based abstractions of molecular substructures. Then search through these representations to find promising combinations that would yield good products.
Typical inputs
Reaction schemas (e.g., amide coupling, SNAr, click chemistry).
Pools of compatible synthons for each reaction role (acids, amines, aryl halides…).
A scoring function that evaluates fragment or feature matches (topology, pharmacophore-type features, etc.) and combines them.
How it works (under the hood)
Two common strategies:
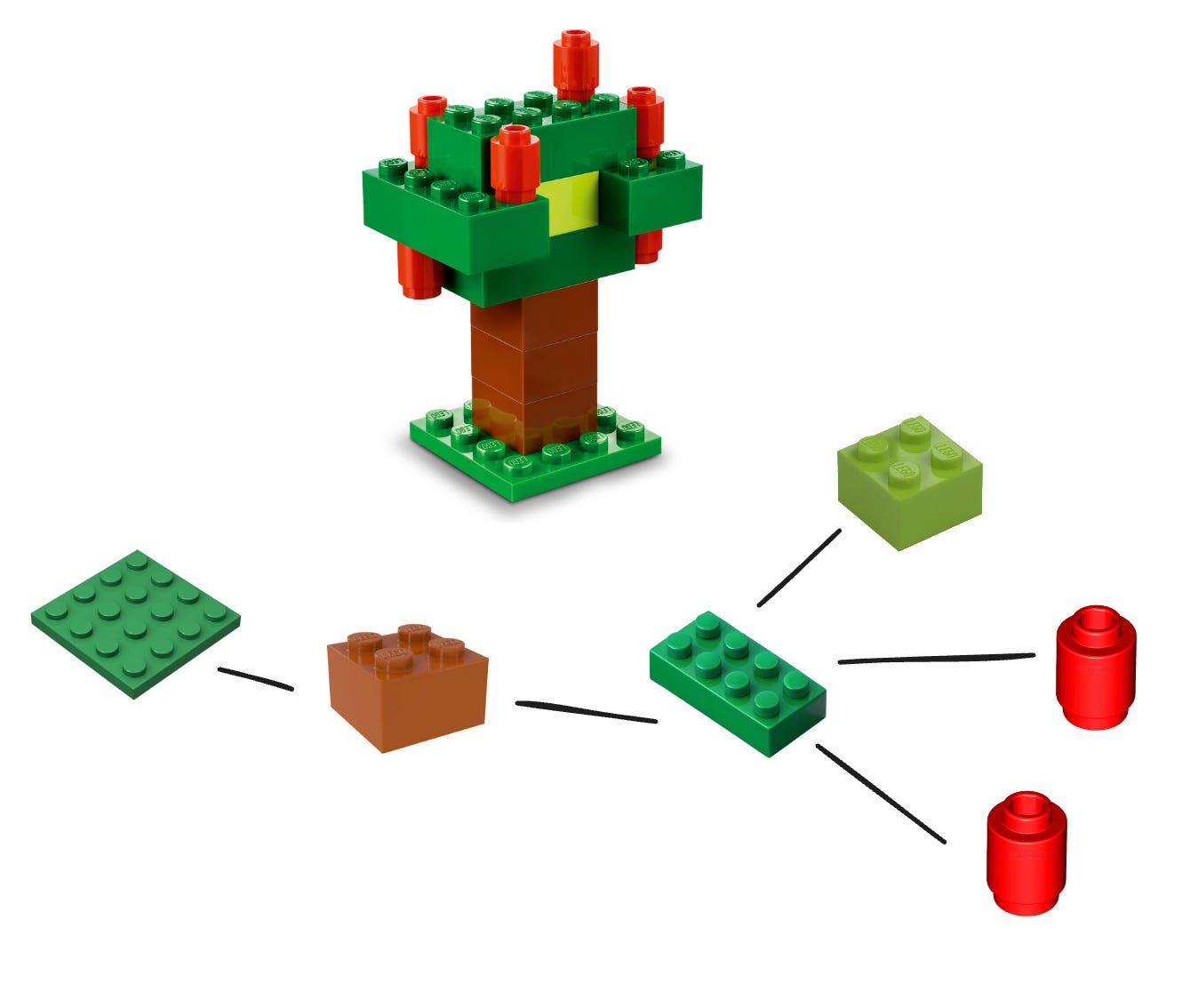
Disassemble & reassemble (e.g., Spacelight, exaScreen, Hyperspace)
Cleave the query molecule into synthons at strategic linkers.
Find similar building blocks for each fragment.
Reconnect the highest-scoring fragment combinations, often relying on the idea that similar parts yield similar products.
Feature-tree matching (e.g., F-Trees)
Represent the molecule as a tree of linked feature nodes (e.g., donor, acceptor, aromatic, hydrophobic, ring presence, spatial volume).
Search for the most similar feature trees, without requiring exact fragment matches.
Both methods pre-index the search space (far smaller than enumerating all products), match compatible parts or features, and only instantiate full molecules for the top-ranked hits (“lazy enumeration”).
Strengths
Scale. Traverses libraries far beyond what you’d explicitly list (billions → trillions) on modest hardware.
Synthesis-aware. Every suggested product corresponds to a plausible reaction with defined parts.
Speed. Fragment indexes are compact; combinatorial search can be near-instant.
Limitations
Constrained space. If a scaffold isn’t reachable by the chosen reactions/building blocks, you won’t find it.
Fragment approximations. Product quality is inferred from parts; whole-molecule effects (3D clashes, stereochemistry, conformational strain) can be missed until instantiation. Not all properties are compositional and additive.
Ranking uncertainty. High-scoring fragment combos don’t always yield the best products after full evaluation.
Prefer enumerative when…
You need exact whole-molecule scores (e.g., feature overlap, 3D shape or electrostatics, substructure, stereo, physics) without any approximation or compromise.
You want to exploit curated, well-understood catalogues, high hit rate, in-stock collections. Compounds you can directly purchase.
Library size is manageable for your compute/storage budget.
Diversity is crucial for you and the combinatorial chemical spaces you have tried have structurally limited or repeating chemotypes. You are unable to find in them what you want.
You want to filter the compounds based on properties, predict ADMET or train QSAR models on the precomputed fingerprint or embedding representations.
Prefer synthon-based when…
You need to explore far beyond today’s enumerated databases for novelty.
You’re mapping vast “what-if” spaces with minimal compute and storage. You can run the search or screening on your own laptop.
It is fine for you that not all of the molecules will be synthesisable and success rate varies based on catalogue.
You are looking for a match in pharmacophoric features or cleaved fragments, not necessarily the most similar analogues.
You want scaffold hops and you are not concerned with subtleties in electrostatic similarity or substructure matching.
A mini-glossary
Enumerated library: A concrete list of molecules (structures known in advance).
Synthon: A fragment with defined attachment points representing a role in a reaction.
Fingerprint: A binary representation of a molecule
Embedding: An AI-based vector representation of a molecule
Reaction schema/template: A generalized reaction pattern that stitches synthons into products.
Lazy enumeration: Generating full product structures only for shortlisted combinations.
ADMET: Absorption, Distribution, Metabolism, Excretion, and Toxicity
QSAR: Quantitative structure–activity relationship